
Elisia’s Story
“When Elisia was born in April 2015 as far as we knew it she was ‘normal’, weighing a healthy 8lb 8oz. She passed all her new-born screening tests and was absolutely gorgeous.
We joined the standard classes and groups making mummy and baby friends at each one. Everything was lovely for the first few months. Then when Elisia was between four and six months a few things started playing on my mind and I wondered why she was not doing as well as the other babies. Everyone kept telling me: ‘all babies are different’, ‘she will get there in the end’ ‘all babies learn at different rates’ etc.
When she reached about seven months old things became more difficult. Taking her to classes and groups became a struggle. Simple questions from other mums upset me, even the standard opening question ‘how old is she?’ made me anxious. I would think they were asking because they had noticed how little she could do. The list of things she should be able to do but couldn’t was now getting long: rolling, sitting unaided or crawling. So, I went to the GP who agreed a referral to a paediatrician was needed.
We had to wait until she was nine months old to get a date for this appointment, so we enjoyed our first Christmas together as a family of three and carried on as usual. We also attended a lot of Elisia’s friend’s first birthday parties. This was difficult – they were all walking or at least standing and walking with a parent whereas we were getting excited as Elisia had just mastered a roll!
When we saw the paediatrician and told him the milestones Elisia had not yet met, he referred us straight away for tests. Bloods were taken that day and an MRI scan booked for the following month. That was such a scary and long day at the hospital for a little girl but she handled it so well. She is a placid and content person.
All Elisia’s tests and MRI scan came back negative. The paediatrician told us he was waiting on the genetics team to get back to him. Everything sounded fine, we made a big sigh of relief. Elisia then had her first birthday in April. She had two parties and we had a little holiday in Butlins – our first family holiday. She couldn’t go on most of the rides but we had a nice time, Elisia absolutely loved the pool.
 When Elisia was thirteen months old we went back to the paediatrician and were given the devastating news that Elisia had a rare chromosome disorder, 5q14.3 deletion. What even is that?! Even the doctor appeared to know practically nothing and couldn’t tell us much about how our little girl would be affected as he hadn’t come across it before. He downloaded the 5Q14.3 deletion leaflet produced by the charity Unique which explained the condition and then sent us on our way feeling frightened and bewildered.
When Elisia was thirteen months old we went back to the paediatrician and were given the devastating news that Elisia had a rare chromosome disorder, 5q14.3 deletion. What even is that?! Even the doctor appeared to know practically nothing and couldn’t tell us much about how our little girl would be affected as he hadn’t come across it before. He downloaded the 5Q14.3 deletion leaflet produced by the charity Unique which explained the condition and then sent us on our way feeling frightened and bewildered.
Reading the leaflet I was in flood of tears… ‘may not walk till much older if at all, may never talk, prone to autism, epilepsy, hypotonia etc’
I was numb. I tried my hardest not to get too upset or stressed because at this point I was five months pregnant with our second child. I knew I needed to be strong for my unborn child and also be there for my little girl who that same day had further blood tests along with Mummy and Daddy to see if one of us had the same chromosome deletion and had passed this to her. We later found out that it was a ‘de novo’ chromosome deletion which means there was no family history of this genetic change and it appeared in Elisia for the first time.
We went to see the genetics team in Winchester a few months later. We had high hopes that at this appointment someone would finally be able to answer our many questions. However, we were handed the same Unique leaflet that we had been given at the paediatrician appointment! We were told that Elisia seemed to be doing quite well (compared to many of her 5q14.3 deletion peers), she was strong so clearly did not have hypotonia which is one of the most common symptoms with her deletion. We did however find out that within her 5q14.3 deletion she was missing an entire gene – MEF2C. We later found out this is a commanding gene and explained many things. She seemed to show more similar traits with people who had an issue with MEF2C rather than just a 5q14.3 deletion.
We then found the MEF2C group on Facebook and I can honestly say I feel like I am now part of a global family. This group is small group – 273 members, so I feel like I know many people quite well. We support each other, pick each other up and no matter the time of day (if Elisia is not sleeping at 2:00am or we are in hospital and I cannot sleep for instance), one of the other group members on an opposite time zone e.g. Australia or America will talk to me, support me and cheer me up.
When Elisia was seventeen months old, her baby brother was born. As I had to have a c-section, for the first twelve weeks we had visitors every day and we had lots of support. Then followed some of the loneliest and darkest days of my life. It was winter and I struggled to leave the house with both babies as I couldn’t carry them at the same time. The visitors became few and far between and the days were long and hard. Then the second birthday invites started to roll in. I felt pleased that Elisia was still being included but also extremely anxious about the fact that she was VERY different to her peers now. She was still a ‘baby’ and they were all clearly ‘toddlers’ running around, jumping, laughing etc, whereas Elisia could not even stand or crawl.
first twelve weeks we had visitors every day and we had lots of support. Then followed some of the loneliest and darkest days of my life. It was winter and I struggled to leave the house with both babies as I couldn’t carry them at the same time. The visitors became few and far between and the days were long and hard. Then the second birthday invites started to roll in. I felt pleased that Elisia was still being included but also extremely anxious about the fact that she was VERY different to her peers now. She was still a ‘baby’ and they were all clearly ‘toddlers’ running around, jumping, laughing etc, whereas Elisia could not even stand or crawl.
When Elisia was a little under two years old her little brother started crawling at just 5 months old. It hit me hard that her little brother had overtaken her developmentally. Elisia had been trying all year to master crawling and he picked it up in a few days. I was honestly depressed but I also felt incredibly guilty that I couldn’t be happy for my son as I was grieving for Elisia. It seemed to come so easy to him with very little effort. Why is life so unfair? to make some children work so hard for what many people take for granted. However, with each passing milestone my son achieved, for instance walking at just 9 months I found it got easier and I could be happy for him. I may never hear my daughter say she loves me and that is the hardest thing with all this. I can cope (well possibly my back can’t) with the physical side of this. But if she never talks, that I think will slowly break my heart.
At 2 ½ years old she got a place at a fantastic special needs nursery attached to a school. She also has lots of equipment now like a stander, a supportive chair and a walker. Most of her specialists now see her at nursery, so our hospital visits have decreased considerably. I know some people do, but I didn’t consider mainstream for long at all. For now, this is the best place for her. Whether this will change I am not sure. Within three months at the nursery she was doing well and started crawling! It’s about a year since then and I still stop whatever I am doing to watch her crawl as I am still overwhelmed and emotional to see it.
I often get the comment ‘well, at least she is so beautiful’ well, yes, she is and it isn’t always easy to identify her disability straight away, especially if she is sat down. Because of this I am continually explaining that she has additional needs when people give her something to hold and she doesn’t reach out for it or they try to talk to her and she looks the other way for example. It is also uncomfortable when she bites her hand, grinds her teeth, stims or jerks. People don’t know how to handle this. Even people close to us often laugh it off, stare at her or say something inappropriate. I don’t want people to walk on egg shells around us but do wish people thought more before speaking and filter whether that comment or action could be deemed upsetting.
Elisia continues to amaze me all the time. She can almost now stand unaided and is trying so hard to pull to stand up. She loves music and water. I often wish I had an easier life. But if that meant giving her up (or indeed giving up on her) I soon stop wishing that. I am blessed to be given my daughter. She is unique, loving and so very special. She puts a smile of my face every day.
We are very grateful to have received support from various wonderful charities and I wish we had found some of them earlier on our journey.
The Sandcastle Trust is an amazing charity. It is a small one but they make such a difference to the families they support. We really appreciated the family day out to see Santa at 4 Kingdoms Adventure Park and Farm that they awarded us through their Sandcastle Santa scheme. The whole family had a lovely day out and the grotto had amazing lights and music which Elisia loves. Christmas is such an expensive time of year, especially when special needs items cost so much more than ‘normal’ presents so we really appreciated being awarded a family day out to see Santa – Thank you to The Sandcastle Trust!

Jamie’s Story
Jamie Tyler was three years old when he died in April 2019 from the rare genetic condition Leigh’s Disease. Jamie’s family were gifted a stay in the CBeebies Land Hotel at Alton Towers by the Sandcastle Trust in his memory. Here Jamie’s mum Sam, 36, from Hereford, explains how much the trip means to them.

Sam said: “I had a normal pregnancy with Jamie, and he was my third child. He seemed to be developing normally until he was five months old. He had been starting to smile and babble, but I noticed he had stopped making noises and was no longer giving us his cheeky grin.
“Then he started to lose interest in food, he no longer wanted to hold his cup or eat his toast, and by his nine month check we couldn’t tick any of the boxes for the milestones he should have been reaching.
“We were referred to hospital in Birmingham, then a specialist centre in Newcastle, and by April 2016, when Jamie was 16 months old, we had a diagnosis of Leigh’s Disease.
“It’s a rare genetic condition that affects only 1 in 40,000 newborns. There are different variations of the disease, but doctors found out that Jamie had the mitochondrial type, which is passed on by the mother. I was tested and they found low levels in my blood, as well as in Jamie’s older siblings Sophie, who’s now 17, and Jack, who’s now seven.
“There is very little information available about what to expect, but we were later told Jamie would continue to become more and more poorly, and his life expectancy was just three years old.
“I was devastated at the diagnosis, I had felt it was something serious but neither me nor Jamie’s dad Barry had ever heard of this. You just think Jamie will get to 10, then he’ll get to 16, then we’ll see after that. But hearing we would only have another two years with him, together as a family, was a huge blow.
“I was already five months pregnant with our fourth child Rebecca by this point, and Barry and I knew that if she had the condition then we would deal with it, but so far she hasn’t shown any signs of being affected.
“Once we knew we didn’t have much time left with Jamie we decided to make as many memories and take as many pictures as we could.
“Barry and I got married with the children as bridesmaids and page boys, and we organised lots of days out for all of us. Jamie and his brother Jack were so close, and they loved watching CBeebies together. Jamie’s movement was limited and he couldn’t speak, but when the show Swashbuckle came on he would move his arm in the pirate salute and we knew he was enjoying it.
“We had planned to go and stay in the Swashbuckle room at the Alton Towers hotel, but sadly Jamie passed away before we could go on the trip.
 “Jack was devastated to lose Jamie, we all were. Jack did so much for Jamie and he never thought of himself or complained, he knew Jamie’s needs came first. Even now, when he looks up to the sky and sees a star shining brightly, he tells me it’s Jamie looking down on us.
“Jack was devastated to lose Jamie, we all were. Jack did so much for Jamie and he never thought of himself or complained, he knew Jamie’s needs came first. Even now, when he looks up to the sky and sees a star shining brightly, he tells me it’s Jamie looking down on us.
“Together with the pandemic it’s been an awful two years and I wanted to give the children something to look forward to. So when I contacted the Sandcastle Trust and they said they would organise the Alton Towers trip for us, and for us to stay in the Swashbuckle room, I was thrilled.
“Jack is so excited, and the first thing he said was that he wanted to make sure Jamie could go too. We all have pillows with a picture of Jamie on them, and Jack has already decided he’ll be packing his pillow so his brother will be with him.
“It’s still so important for us as a family to create memories together, given what we’ve all gone through. Life is too short to wait and I firmly believe in having fun, staying positive and moving forward.
“This trip will help us all to do that, and thank you so much to the Sandcastle Trust for giving my family something really special to look forward to.”
The Family’s trip to Alton Towers




`

Kennedy’s Story
Kennedy Mercer was five months old when a rare genetic condition claimed the life of her twin brother Karter. Kennedy has the same condition, Spinal Muscular Atrophy Type 1.
Both twins were diagnosed with the disease before turning six-months-old. Babies with it usually die within two years, often as a result of serious breathing difficulties, but smiley seven-year-old Kennedy has astounded doctors with her fight for life.

SMA Type 1 is a neuromuscular disorder which causes severe muscle weakness and progressive loss of movement. Babies with it are usually unable to sit upright unsupported or lift their head. They are prone to chest infections and have difficulty swallowing. All muscles are affected including the heart and lungs. Intelligence isn’t affected and babies are often bright, alert and responsive.
Karter and Kennedy were born premature in September 2012 after mum Jeannette, was diagnosed with preeclampsia. “The twins were well fought for, it was our eighth round of privately-funded IVF,” explains dad Paul, an HR executive from Grays, Essex.
“Other than arriving a little premature, everything was going well. Karter was two months old when I noticed he was crying through his feed and struggling to swallow. I noticed he hadn’t moved his legs in some time. We didn’t think for a minute he had a life-threatening illness but we wanted to get him checked so we took him to our local hospital in Basildon. He was given lots of tests and we paid privately for him to have an MRI scan in London. Everything came back clear.”
Doctors ordered one final test to check the nerve endings in Karter’s spine and it was then he was diagnosed with SMA Type 1.
“We were told, subject to blood tests, that doctors were 99 per cent certain he had SMA Type 1,” Paul explains. “They said there was nothing they could do for him and it was unlikely he’d make his first birthday. It was a life-changing moment. You start grieving right then when you are told your baby will die, even though he is still there, looking at you, smiling at you. You grieve for things you now know will never happen, such as seeing him kick a football, swim in the sea or run around the garden. I’d never heard of it before, I knew nothing at all about SMA.”
SMA is a recessive genetic disorder, meaning both parents must carry a faulty copy of the SMN1 gene for it to occur. If both parents are carriers – and research suggests one in 40 of us are – there is a one in four chance in each pregnancy that the baby will be born with SMA. Most people do not know they are a carrier until they have a child with the condition.
“We asked whether Kennedy might also have it and we were told that it was a possibility but the odds were in our favour,” Paul says. “She had a 25 per cent chance, the same as her brother. Kennedy’s blood tests were carried out in November 2012 but we asked not to be given the results until after Christmas. A few days afterwards, a local consultant turned up at our front door with a family liaison officer. We knew instantly she must have it, she was only four months old.”
The twins’ parents decided they’d give their children the best lives they possibly could – no matter how short. “We could sit at home crying and wondering why this happened to us,” Paul says. “Or we could do our best to give them the best quality of life we could.”
Sadly, Karter died of respiratory failure in February 2013 aged five months.
“He was in bed with me and I woke up about 2am to do his feed but he was cold and unresponsive,” Paul says. “I tried to resuscitate him. An ambulance arrived and paramedics took over, but we couldn’t get him breathing. I made a promise to him that night that we would ensure Kennedy had the best life possible. For a lot of people, it’s about keeping their child alive as long as possible but over time we realised that was not the be all and end all. We focus on making memories for her and making sure she has a brilliant life.
 “I started doing more research and I flew to the US to meet one of the world’s leading specialists on palliative care for children with SMA. We had a challenge to get her the treatment we wanted but we pushed for her to have a drug called Spinraza. She started getting it when she was five and it’s made a huge difference to her life. If Karter had been given it at birth, he’d still be here now. It hasn’t been approved for all types of the disease, so we are lucky she got the drug. If it is given to newborns, SMA Type 1 isn’t necessarily seen as a terminal illness any more.
“I started doing more research and I flew to the US to meet one of the world’s leading specialists on palliative care for children with SMA. We had a challenge to get her the treatment we wanted but we pushed for her to have a drug called Spinraza. She started getting it when she was five and it’s made a huge difference to her life. If Karter had been given it at birth, he’d still be here now. It hasn’t been approved for all types of the disease, so we are lucky she got the drug. If it is given to newborns, SMA Type 1 isn’t necessarily seen as a terminal illness any more.
“With Kennedy, it has helped her to speak. She can sing songs. She loves Let It Go from Frozen. She has been able to express herself much better and it’s made her far more independent. She can draw, paint, go to school, go on days out and drive her own powered wheelchair.”
Kennedy is doing extremely well. She is home-schooled and goes into a local mainstream school on an ad hoc basis. “It is difficult to go regularly as there are so many bugs around and a simple cold could be life-threatening,” Paul explains.
Day to day, Kennedy’s parents ensure her airways are kept clear, which involves using a suction device every hour during the day. At night, she breathes through a ventilator. Kennedy is unable to eat or drink and all food is administered via a peg in her tummy. This allows food to go directly to her stomach, bypassing the most and throat.
She has regular checks at London’s Great Ormond Street Hospital, where she goes for her Spinraza medication, as well as weekly sessions with a physiotherapist.
Mum Jeannette gave up her job after the twins were diagnosed and is now Kennedy’s main carer. Paul works from home so he too can care for his daughter.

“After what happened with Karter, we decided to always keep her close to us,” he says. “She sleeps in bed with one of us. We don’t have any night care, but we take it in shifts so one of us is always by her side. Her brain works normally so sometimes she wakes in the night and wants to turn, but she can’t manage it. One of us has to physically turn her.”
“Most children of her age with SMA need a ventilator to help them breathe at all times, so Kennedy is very rare,” he explains. “When she is awake she can breathe by herself. On the whole she is doing very well.”
“She will never be able to walk, she’ll never sit up independently and she can’t play with her friends in a traditional sense. But she is one of the happiest children on the planet. She loves Disney and dressing up, she has every Princess outfit you could think of. She loves Barbie and playing with dolls just like many other little girls. She is at her best when she is surrounded by children her own age. She loves spending time outside in the garden, she loves swimming.
“We want to create as many memories as we can for Kennedy. She’s flown over 15 times, she’s been to Disneyland, she has swam with dolphins, fed giraffes, she’s been to the cinema and theatre. We want to give her as much independence as possible.”
The family have been supported by The Sandcastle Trust.
“The charity recently arranged for us to stay in the Cbeebies Hotel at Alton Towers which Kennedy loved,” Paul says. “She loved all the rides and meeting her favourite characters from the telly. The Sandcastle Trust do fantastic work with adults and children to try and create lasting memories.”


In 2016, Paul and Jeannette had a second daughter Marli, now aged three, after having pre-implantation genetic diagnosis (PGD) at Guy’s Hospital in London. This is a type of IVF which meant doctors were able to rule out the possibility of their baby having SMA Type 1.
“The doctors had to take DNA from all the family but the doctors could tell us with 99 per cent certainty before putting the embryo back in whether it was going to be SMA positive, an SMA carrier or have no SMA at all. We know Marli is a carrier, but we knew before she was born that she wouldn’t have the disease. The two sisters are as close as they come. The love they have for each other is second to none.”
Jeanette also has three elder children from a previous relationship, Courtney, 22, Kelsey, 18 – a regular on The Only Way Is Essex – and Bill, aged 19.
“We are not your average family but we are a very happy one,” Paul says. “Of course, I have spells when I 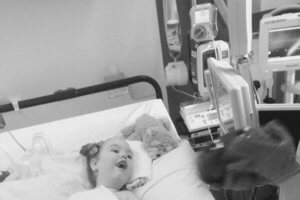 worry about the future. Most parents who look after a child with a rare genetic condition sometimes worry about what tomorrow might bring, especially if you are dealing with a condition where the life expectancy is two years or less. We know we could lose her at any time.
worry about the future. Most parents who look after a child with a rare genetic condition sometimes worry about what tomorrow might bring, especially if you are dealing with a condition where the life expectancy is two years or less. We know we could lose her at any time.
“We try not to think too far ahead any more, we live in the present. We try to make sure that every day is the best it could have been. I look in the mirror every night and question whether I have done the best for her that day. I feel blessed to have Kennedy. I may have saved her life many times but she has also saved me. My life is completely different now. I don’t worry about financial rewards, eating in nice restaurants or buying nice clothes. What matters to me is seeing my daughter happy. Of course, life can be hard sometimes, but I wouldn’t change her for the world.”

Tilly’s Story
“Our little girl Matilda, Tilly, was diagnosed with Rett Syndrome in March 2018.
Tilly had developed normally until she was one, with no concerns whatsoever, but then started to gradually lose her skills.
Rett is a rare genetic neuro-developmental condition affecting mainly girls leaving them profoundly disabled.
Rett means Tilly cannot walk, talk, use her hands, transition between postures, and needs 24-hour life long care for every single need.
 To say the news was devastating is an understatement and it has had a huge impact on our family – me, Tilly’s dad and Tilly also has a three and a half year old sister Thea, who we try and ensure is unaffected as much as possible by the diagnosis.
To say the news was devastating is an understatement and it has had a huge impact on our family – me, Tilly’s dad and Tilly also has a three and a half year old sister Thea, who we try and ensure is unaffected as much as possible by the diagnosis.
During the first few months we were left reeling, and bad news after bad news kept on coming at her various appointments; she may need a feeding tube, she might not walk and intellectually, she may not exceed the age of a 2 month old.
When Tilly started to have seizures and tremors things became really tough for our family.
So, when we discovered The Sandcastle Trust after looking through a handout given to us to find funding for a pushchair, we decided to contact them.
Through the Sandcastle Trust we were able to have a fantastic break in North Yorkshire, a day after a really important meeting to discuss Tilly’s educational needs.
We stayed in a brilliant caravan and had three days just us as a family, taking time to process what had happened, just get away and make it fun for Tilly and Thea.
They both loved it; we went to the children’s disco, the beach, the swimming pool which was amazing and everything was geared towards the children. It was a holiday we would never have taken and we loved it that much I am sure we will go back.
Tilly’s symptoms are now manageable, but every time we go anywhere we need to blend all her food, take all her medication, and equipment, and make sure she has everything she needs so she doesn’t get distressed or upset.
The place we stayed in was huge and so it meant that we could take along everything we needed.
A year on from her diagnosis, Tilly is thriving. Although Rett has had a significant impact physically, there is nothing to suggest that she is not the same age mentally as other two and a half year olds.
Going on this break took us away from the seriousness of everything, helped us gain a sense of hope and peace restored, which gave us the strength to carry on. The Sandcastle Trust made that possible for us and I will be forever grateful.
What The Sandcastle Trust do is give you space to be able to get away as a family and breathe. That is what we needed to be able to step back into the uncertainty of life and what it now means for us.
Thank you Sandcastle Trust for supporting us it meant and means the world to us.”


Jacob’s Story
When their baby son Jacob was diagnosed with a life-limiting genetic disorder parents Gemma and Kevin Rimmer knew their world had changed forever.
Gemma, from Liverpool, says: “We realised our gorgeous boy would need 24-hour care. I had to give up work as a hairdresser. Kevin had to leave a good job on the railways for something more local. Alder Hey Hospital became our second home.
 “For the first few months of Jacob’s life his older brother Joel was passed from one family member to the next while we sat by Jacob’s hospital bed.”
“For the first few months of Jacob’s life his older brother Joel was passed from one family member to the next while we sat by Jacob’s hospital bed.”
The idea of a holiday or a day out seemed completely unattainable.
Gemma says: “Kevin became a taxi driver so he could be near us in an emergency. Our income reduced considerably and all our spare time was spent entirely on the boys.
“So when the Sandcastle Trust offered us a break it was absolutely magical to be able to take them away for the night.”
The charity gave the family a trip to Alton Towers so Jacob, now three, and Joel, six, could visit CBeebies Land there.
Gemma says: “Seeing the excited looks on the boys’ faces at the CBeebies Live shows was something I will always remember. We stayed in the Mr Tumble room which is Joel’s favourite.”
When newborn Jacob was just two days old he suddenly stopped breathing. He was rushed by ambulance to Alder Hey Hospital in Liverpool and hooked up to a life-support machine. He remained in a coma while doctors urgently tried to figure out the cause.
Lumbar puncture tests showed his glycemic levels were high. Doctors diagnosed Nonketotic Hyperglycinemia (NKH).
Gemma says: “They told us Jacob would need a lifetime of care, he could have as many as 24 seizures a day. They told us to summon our family to say goodbye and suggested we switch his life support machine off.
“We knew we wanted to keep him alive. We felt that if he had wanted to die he would have gone before the ambulance arrived at our house.”
Jacob deified doctors’ expectations and eventually started feeding from a bottle. At three months the family were allowed to take him home. He now smiles and giggles, can say a few words, loves watching CBeebies and is learning to use a ‘switch’ toy to increase his motor skills.
NKH causes epilepsy, global mental delay and stomach problems and Jacob also has cerebral palsy. The family continue to spend long periods in hospital.
Gemma and Kevin have become used to the routine of caring for him.
Gemma says: “Jacob rarely sleeps so we take turns being up in the night. He is fed through a peg in his stomach. His first meal is at 6am. I give him all his medication at 8am while I’m getting Joel ready for school. Then Jacob will have a feed every four hours, four times a day.
“Nappy changing is always a fight, he rolls away from us.
She adds: “Someone said to me the other day “you don’t look like you any more”. I used to wear make-up and always have my hair done but I just don’t have the time now. I can’t remember the last time Kevin and I went out as a couple.”
The Sandcastle Trust has also helped Jacob and Joel visit Father Christmas in a local grotto two years in a row.
Gemma says: “With Jacob we take each day as a blessing so it’s always bittersweet to visit Santa, it marks another year he has survived.”



Cally’s Story
Mum Cally Benvie, 30, from Broughty Ferry, Dundee, is a wheelchair user after being diagnosed with the degenerative genetic disorder Friedreich’s Ataxia as a teenager.
She enjoyed a blissful weekend Center Parcs in Whinfell Forest, Penrith, with partner Alan Moffatt, 33, an NHS porter, son Jay, 10, mum Jill, a nurse, and her mum’s partner George.
The trip, organised by The Sandcastle Trust, enabled her family to be just that, a family and not have to worry about Cally’s disability for the first time in a while.
Chatty Cally says: “It’s not often I’m speechless but the holiday was so incredible I am lost for words. It was the first family break we have had since I had to use the wheelchair full time.
“I’m crying as I say this. A lot of Alan’s attention has to be on me but going away with my mum meant Alan and Jay could do whatever they wanted without having to worry about me. Jay could come first.
“It was fantastic as a mum to see how much enjoyment my son got out of the weekend. It felt like we weren’t focused totally on me having a disability we could just focus on being a family. It was magical.”
a disability we could just focus on being a family. It was magical.”
Friedreich Ataxia causes progressive nervous system damage and impaired muscle coordination that worsens over time. It was Cally’s dad who first spotted there might be something wrong with his daughter’s movement.
She explains: “Dad lives in Wales so I didn’t see him regularly. I was visiting him and he asked me ‘What’s wrong with your legs? You’re walking strangely.’
“I hadn’t noticed and told him not to worry but he was so concerned he insisted I saw a doctor when I got back to Scotland, which I did.”
Suspecting a brain tumour, Cally’s GP sent her for an MRI scan straight away. The results were negative so the doctor sent Cally’s blood for genetic testing. She recalls: “I thought I might have something I could take a tablet for and it would go away, something manageable. But it wasn’t.
“The doctor told me it was Freidreich Ataxia then gave me a leaflet he had printed from the internet and a website address.
“I’d never heard of it and didn’t think too much of it. I had my lunch and went back to work.”
Cally Googled the condition. She says: “That was a mistake. Google had me dying in a couple of years, it still didn’t sink in. My mum was doing the same thing and was horrified. It was when I told my mum and she got upset that I realised it was more serious.”
Cally had only just met Alan at the time. She recalls: “19 is an awkward age to be told you have a life-changing condition. Alan could have walked away then and there but he’s stuck with me. He’s been a great support.
“I don’t think I would be doing as well without him. He’s a really good listener. I found a good one there.”
The first time Cally remembers FA impacting her came a couple of months after her diagnosis. She says: “I met Alan on a Friday after work. The doorman at the bar where we were going said: ‘You’re not coming in, you’re too drunk.
“Then he said to Alan: ‘You need to get her up the road pal. She’s had too much.’
“I explained I had come straight from work and hadn’t had a drink. I was angry he wouldn’t speak to me but over my head to Alan. He wouldn’t let me in. He said I was making it up. It would have been a bit of a thing to make up.
“I’ve never been back there.”
“Now my balance and coordination aren’t great and my speech can be quite slurred. It’s got worse over the years but because I use the wheelchair all the time now everyone knows I’m not drunk.”
Jay was born in 2011 and the family have learned to live with Cally’s condition. Cally says: “Jay is very understanding and a big help around the house when Alan’s at work, small things like getting something from the fridge or closing the blinds. He is always happy to help me.
“When he was younger he would just tell his mates ‘my mum’s a bit wobbly’ and they would carry on playing. I’ve found children are a lot more accepting than adults.”
She says: “I can’t walk by myself any more but we get by. We live in an adapted house with a ramp. I use a wheelchair all the time in the house.”
“My condition is progressive but I am quite a positive person. Like everyone you have the odd day when you’re not doing so well.”
Cally was furloughed from her office job during lockdown. Researching an accessible holiday to cheer the family up she stumbled across the Sandcastle Trust. She recalls: “Most charities are based around children, certainly ones for genetic disorders. But my disorder impacts my family.
“I was tearing my hair out with homeschooling. It was so lovely and reassuring to talk to Danielle from the Trust, she understood.
“She suggested Center Parcs and kindly agreed that Mum and George could come too.” 
The family travelled in July 2021 at the start of Jay’s school holiday.
Cally says: “After Covid it was amazing to be away from home. It was like a dream.
“My mum and I don’t get much time together but we got to have afternoon tea. The boys went swimming without the additional pressure of me in the wheelchair.
“Alan and Jay are very good, they would never say I’m an issue but they could have quality time together.
“We stayed in a wheelchair-adapted lodge. All the counters were at my level. The shower had a shower chair, my bed was a hospital bed. We didn’t have the hassle of bringing any additional disability things with us.
“It was the most incredible weekend. If I was a millionaire I would be there every week.
“Jay loved all the activities, there’s wildlife, tennis, badminton, shops and restaurants. But his fave things were the nights we were back in the lodge and we played board games and ate pizza.
“We would recommend it to anyone. The staff couldn’t do enough for us. They took my disability seriously but it felt really, really safe and still like a holiday.
“Words aren’t enough to explain how grateful we are to everyone at Sandcastle.”

Maisie’s Story
Sixteen-year-old Maisie Doswell suffers from such debilitating headaches she has to spend most of the day lying flat on her bed. Maisie has the rare genetic disorder Myhre Syndrome, of which there are only thought to be 30-40 reported cases worldwide.
From the age of seven she has suffered from excruciating headaches – caused by a build-up of fluid in her  brain. She has had a shunt fitted in her lumbar spine to drain the fluid. It drains when she is upright and is causes terrible headaches on draining. Lying flat is the only way Maisie can get any relief from the pain.
brain. She has had a shunt fitted in her lumbar spine to drain the fluid. It drains when she is upright and is causes terrible headaches on draining. Lying flat is the only way Maisie can get any relief from the pain.
Mum Karey Mason explains: “At the moment she is just a teenager in a bed unable to get up for more than half an hour because of the headaches. Painkillers don’t work. She begs me for help, what can I do?”
When Maisie Doswell was born in 2003, doctors immediately picked up at birth that she could have a genetic disorder.
“My pregnancy was perfect,” Karey recalls. “Then, as soon as I’d given birth, the room filled with doctors. Maisie had low set ears, sunset eyes, which means she couldn’t look upwards, and a Palmar crease – which is a single crease across the palm of the hand. I was so tired I didn’t take anything in. At first, the doctors thought she had Down’s Syndrome, but that test came back negative. She spent four days in hospital and then we went home and we waited for a paediatric referral.”
Karey, 39, from Rye, East Sussex and her then partner Ivan Doswell, noticed that Maisie was slow to meet her milestones, particularly slower to crawl, walk and talk.
At four-years-old Maisie said her legs were hurting and Karey had to rush out and buy a buggy for her to use. Then at the age of seven Maisie went through an early puberty. At the same time, she suffered excruciating headaches.
Maisie was referred to Kings College Hospital, where they diagnosed intercranial hypertension – caused by a build-up of fluid in the brain. She had a shunt inserted into her brain and since then has been re-shunted over ten times.
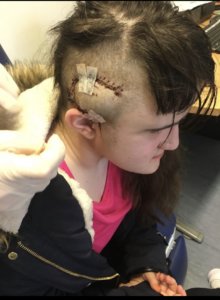 Since the age of seven Maisie’s health has deteriorated. She has lost the hearing in one ear and due to a lack of blood supply at the back of her left eye she has lost the total vision in that eye. Her vision in her right eye isn’t 100% and doctors are battling to save the sight. She now has a lumboperitoneal shunt which drains fluid from her brain into her abdomen. If the fluid in her brain isn’t drained sufficiently she risks losing the remaining sight in her right eye.
Since the age of seven Maisie’s health has deteriorated. She has lost the hearing in one ear and due to a lack of blood supply at the back of her left eye she has lost the total vision in that eye. Her vision in her right eye isn’t 100% and doctors are battling to save the sight. She now has a lumboperitoneal shunt which drains fluid from her brain into her abdomen. If the fluid in her brain isn’t drained sufficiently she risks losing the remaining sight in her right eye.
Maisie had been referred to a geneticist who suspected she had Myhre Syndrome. It took two and a half years for genetic testing to come back positive.
Myhre Syndrome is an extremely rare genetic disorder, with only 30-40 cases reported worldwide. It is caused by a genetic misprint in the SMAD4 gene on chromosome 18. The majority of cases occur spontaneously at conception, as was the case for Maisie. Myhre Syndrome affects children differently. It can cause respiratory and cardiac problems, delayed development, loss of hearing and vision. Each person with Myhre Syndrome have a 50/50 chance of passing it on to their children.
“At least now we knew what the reason was,” Karey, who is Maisie’s carer, explains. “However, not a lot is known about the condition. Maisie looks like a normal teenager, although she is just 4ft 6” and has size one feet, but it has affected her life massively. She has gone from being relatively healthy to progressively getting worse.
“Maisie hasn’t been at school for almost four years. She has learning difficulties and is academically and socially behind other children. It hurts her head to do anything other than lie flat. It feels like the worst hangover headache. She doesn’t want to go blind, so she must have the drain in to relieve the pressure build up. When she is upright or walking around the fluid drains and that is excruciating. Maisie won’t talk about what she is going through. She loves to knit and listen to pop music.”

Karey adds: “If the doctors set the shunt higher, Maisie wouldn’t have the headaches, but her eyes won’t cope, and she would go blind. It is a horrible option to give a child. Either go blind or live with excruciating headaches. What is scary is that a couple of children with Myhre have passed away.”
In 2009 Karey married Darren Mason, 46, a fencer and coastguard. The couple have two healthy children, Charlie, seven and Lily, six.
“It is a strain on the whole family as we’re not able to go out with Maisie,” Karey explains. “Every day is difficult, we just have to take it in our stride. It is a worry what will happen in the future. It is a scary place to be.”
Maisie and her family have had support from the Sandcastle Trust, “The Sandcastle Trust has been amazing,” Karey explains: “A day out is a struggle for us but for other children it is a stress-free experience. The Sandcastle Trust is there to help. We had a lovely Christmas day out to Bedgebury arranged by the charity. They also organised for Maisie and our family to have a wonderful weekend in London. We stayed in a central hotel and visited The London Dungeon and Shrek’s Adventure – Maisie was so excited!
“Without The Sandcastle Trust, the world would be a bleaker place for families affected by a rare genetic condition.”


Tayen’s Story
Tayen Gilbert, seven years old, is funny, clever and crafty. She knows just how to keep her family on their toes.
Tayen, has Neurofibromatosis (NF1), which causes tumours to grow along her nerves. As a result of her tumours, Tayen is fully blind and is brain damaged. She also has complex epilepsy and has hydrocephalus – swelling of the brain due to excess fluid.
Despite everything, she loves telling jokes, tobogganing, audio books and playing with Lego Duplo.
Her mum, Kali, 37, says: “Tayen keeps us all going. She is just so loveable. Anyone who meets her is so taken with her.”
 The Sandcastle Trust arranged for Tayen and her family to go on a special train ride at Christmas – it gave them a rare opportunity to enjoy the build up to the festive period in a safe environment where Tayen would be comfortable.
The Sandcastle Trust arranged for Tayen and her family to go on a special train ride at Christmas – it gave them a rare opportunity to enjoy the build up to the festive period in a safe environment where Tayen would be comfortable.
Kali continues, “In some areas Tayen is age appropriate, but in other areas she is naturally a little behind. It is very hard to pick apart what is going on inside her brain because of all the damage the tumours have done in there.
“We always used to do day trips with the boys, but it has become increasingly difficult managing her needs out in the community.
“Tayen’s mobility is now really poor, because of all the chemotherapy she has undergone and the tumour growth. Her mind is very active, but her body lets her down. Loud noises worry her, if it’s windy her hearing is affected and she has regular seizures.
“When she gets seizures her behaviour changes because she is in constant pain. She has a wheelchair buggy because her legs give way. It is really frustrating for her.”
Kali has three older sons, Matthew, 14, Connor, 12, Lucus, 12, and is married to Kevin, 40 a lorry driver. The family live in Bridgewater, Somerset.
The family used to have an annual tradition of doing a festive outing but since Tayen’s condition became more challenging they had found it harder to get out into the community.
Kali applied to The Sandcastle Trust for a family trip on the Dartmoor and Weardale Railways Train to Christmas Town. It’s a festive train ride that she knew would be the perfect outing for her sons and daughter.
She explains: “The whole application to The Sandcastle Trust was very quick and easy. I filled in a form in 15 minutes. It was completely stress free.
“We were so grateful that we were able to do a trip as a family so close to Christmas. That really helped with the magic. Tayen was still on chemo so we really didn’t know where we were going to be.
Christmas. That really helped with the magic. Tayen was still on chemo so we really didn’t know where we were going to be.
“The trip itself was wonderful. All my little family in the same carriage which really helped with Tayen’s anxiety. We were able to enjoy the moment, the hot chocolate and the singing. It was lovely.
“Because you are contained in the carriage, I knew Tayen would be comfortable and that the boys would want to go too.
“Sometimes Tayen’s anxiety is so bad I can’t get out of the house. So as a family we are split up most of the time.
“It affects everything really – our sons have grown up missing out on a lot despite all our best efforts. Take parents evening for example – I can’t always get out of the house if Tayen’s anxiety is bad.”
Kali, who was a teaching assistant at a special needs school, says: “I had to give up work to be there for Tayen whenever she needed me.
“She is pretty independent and has chores around the house just like her brothers. We encourage her constantly and love the fact she is inquisitive and wants to try things.
“We will always be so grateful to The Sandcastle Trust for giving our family such a wonderful opportunity. We created memories with all of the children which is so special given the uncertainty of the future.”

Tehyah’s Story
“On 21st July 2010 a little angel was born, my granddaughter, Tehyah. Seemingly healthy, our little princess Tehyah went through life being a gorgeous, cheeky little madam!

On the 11th March 2014 Tehyah went skipping off to nursery laughing. However, at dinner time we had a call to say she was unwell and we should come and collect her. The next morning, my daughter Sam, Tehyah’s Mum, phoned me as she thought Tehyah had suffered a stroke. I just didn’t think this was possible as Tehyah was only 3. I rushed round to my daughter’s house and Sam was right, Tehyah certainly looked like she had suffered a stroke. She was rushed to A&E at Oldham Royal Hospital. They checked her and said it looked like Bell’s Palsy and sent her home with a course of steroids. After four days Tehyah became so sick that Sam took her to A&E again. She was monitored and she spent a few weeks in Oldham Royal. Scans showed nothing and the consultants did not think there was anything seriously wrong. Soon after that they were ready to discharge Tehyah but decided to send her to Manchester Children’s Hospital first for a full MRI scan, just to make sure.
Tehyah was transferred in an ambulance and a full MRI scan was carried out revealing that Tehyah had suffered multiple strokes which had damaged her brain, that the blood vessels to her brain were not all connected, that her vessels in her neck were narrowing and that she had an enlarged heart and kidney problems. Our family were told that our little princess needed an operation or she would die – even with the operation she may still not make it. The hospital were baffled by what was happening 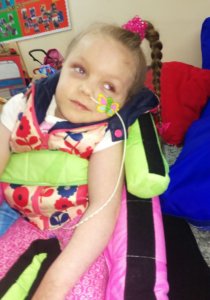
and so needed to contact surgeons all over the world to give Tehyah a chance. Amazingly at this point Tehyah was still running around, eating, drinking, speaking and playing. Tehyah was taken to theatre on the 11th April 2014. She went through ten and half hours of life saving brain surgery. Tehyah woke from this surgery asking for water and she was then transferred to a different ward where she was able to eat, laugh and talk. Eight days later, her condition began to deteriorate and another scan showed brain swelling and fluid on the brain. That night, she was rushed to theatre again to have a drain put in her brain. Three hours later, she was back into theatre as the fluid was building up so much. They removed a part of her skull to allow for the swelling. She was then taken into intensive care, put into an induced coma and on a ventilator for weeks. During this time, expecting the worst, we baptised her. Tehyah fought very hard and eventually she was slowly brought out of the coma and was breathing on her own. At this point we discovered that despite it being on her records that she was seriously allergic to antibiotics, whilst in theatre Tehyah was given a dose of antibiotics that sent her into anaphylactic shock but the surgeons managed to save her. Yet again, after the surgery when she was in intensive care, Tehyah was incorrectly given antibiotics and had to be resusitated.
This resulted in serious brain damage and from this day she never talked, walked or ate again. We were told that she would have no quality of life. On 26th June 2014, Tehyah was given two weeks to live and we took her home. She is still here nearly four years later, thank God! Tehyah now communicates by smiling and laughing and makes sounds when you talk to her and to let you know she’s there. She also attends a very complex needs school called Kingfisher which is amazing and she loves every minute in the school. She has 24 hour round the clock care and is loved by everyone who meets her.
Genetic tests were carried our about a year after Tehyah was discharged from hospital to try and find out what caused all of this to happen. They have now diagnosed her with one of the rarest conditions in the world GACI, Generalised Arterial Calcification in Infancy, which is a life limiting, cruel, horrible condition. It results in the abnormal accumulation of the mineral calcium (calcification) in the walls of the blood vessels that carry blood from the heart. We had Tehyahs older sister Aaliyah and younger brother Dantay tested who at the time were aged 6 years and 4 years. We were absolutely heartbroken and shocked to find out that Dantay has exactly the same condition as Tehyah and that Aaliyah is a carrier of the condition. Dantay is on a medication now to try and prevent the calcification spreading and has regular scans and tests to monitor the progression of the condition.

We are devastated and heartbroken at what has happened to our family and I cannot believe what my daughter, her partner and their beautiful children have gone through and what they will have to face in the years to come. Life has never been the same since that day in March 2014 and will never be the same again, to have known Tehyah from the minute she was born until everything about her was taken away is just unbearable. Family life is difficult as it’s not easy to get out and about now due to all the equipment needed to even go to the cinema, shops, anywhere is a struggle. Tehyah cannot be left unattended not even for a minute, she is doubly incontinent, tube fed, ventilated, on a huge amount of medication and cannot move on her own.
However, we have been lucky to have amazing people around us who have helped us fundraise for the vital equipment that she needs and we have had some incredible support from charities, there are so many people who help and care. We are truly grateful to The Sandcastle Trust for trying to help make life a little more bearable – they provided us with a lovely day out to visit Santa at Chester Zoo this year and we are all looking forward to the very much needed family short break away they have arranged at Brick Farm Cottages next year.”
Debra Walker (Tehyah’s Grandma)





Isla’s Story
“Growing up I never wanted a career all I wanted was to be a mummy.
I met my partner Peter in 2010, I was 25 Peter 28 and we decided to start a family. Unfortunately I suffered two missed miscarriages before becoming pregnant again. I had another scare at 8 weeks but an emergency scan revealed a lovely heartbeat! I absolutely loved being pregnant.
We found out we were having a girl and we knew from then she would be called Isla-Rose.
Isla-Rose was born 14th April 2014. She was perfect.

The first few weeks were spent holding her and watching her for hours. I didn’t get bored of it. Daddy would go to work and come home to find us still cuddled on the sofa. She was growing into a little character and made me laugh every day.
24th August 2014 was a normal summer’s day. Isla was 4 ½ months old. I was getting Isla ready to go out but when I picked her up I noticed she was twitching down her left side, it looked like a trapped nerve so I tried moving her arm. It wouldn’t move. Her face then started dropping. ‘She’s having a stroke’ I said to my friend. I rang 999 and explained what was happening. I started sobbing, my friend said ‘don’t cry Rachael you must be strong for Isla she needs her Mummy to be strong’.
We were blue lighted to hospital and spent five hours in resus while the doctors tried to work out what was happening.
Daddy was on his way, when he arrived and saw his 4 month old baby hooked up to machines with IV lines everywhere he broke.
They managed to stop the twitching and we spent a few days in hospital. Isla recovered and was back to her cheeky self. We were told it was a febrile convulsion. We went home relieved it was all over.
Two weeks later we were out for dinner with family. Isla went quiet and then the twitch appeared. It then took her whole left side. She struggled to breathe. We rang 999 again and spent another night battling in resus to get her back. Her heart rate would hit 200, her oxygen dropped to 80s. They resuscitated her and in the end put her into a coma. We knew then something wasn’t right and more needed to be done.
Isla spent 48 hours on life support but recovered within a day! She shocked us all but still we had no answers as to why. All her tests came back normal.
When we got home we kept an eye on her but it happened again six weeks later. This was to be our story – every six weeks or less Isla would go into a status seizure. Her shortest seizure being 40 minutes her longest, hours!
In total Isla has been on life support four times and blue lighted to hospital seventeen times. I’ve learnt how to resuscitate her and give her emergency medication. Isla was put on three epilepsy medications with no effect. We have to travel everywhere with oxygen.
 Then in January 2015 we were told that Isla has Dravet Syndrome. A rare genetic epilepsy that affects 1 in 40,000. We looked up the condition on the internet where it was described as ‘a catastrophic epilepsy’. We read that children lose their abilities as they grow older and many don’t survive the long, dangerous seizures the syndrome causes.
Then in January 2015 we were told that Isla has Dravet Syndrome. A rare genetic epilepsy that affects 1 in 40,000. We looked up the condition on the internet where it was described as ‘a catastrophic epilepsy’. We read that children lose their abilities as they grow older and many don’t survive the long, dangerous seizures the syndrome causes.
We were devastated, it wasn’t meant to be like this. We had battled hard to get her and now she was given this life. I felt she deserved more from this world.
On the surface Isla looked like any normal child but when Dravet hit, it hit hard. I learnt quickly to be organised and to know all the answers that the doctors needed from me in times of emergencies.
We had nearly made it through the first year of Dravet, which is apparently the most dangerous. But then, in July 2015, I was at a friend’s house and Isla was playing with her little friend, laughing. I noticed a change in Islas tone. I shouted at my friend to grab the oxygen.
The seizure came with such force I struggled to keep her alive. The ambulance was on its way but I knew I needed to save her myself. I kept Isla alive for 40 minutes while I waited for the ambulance. The medicines were given and now my job was to keep her breathing. I lost count of how many times I got Isla breathing again that evening. We lay on my friend’s living room floor while doing lifesaving treatment.
When we arrived in resus they took one look at her and put her straight into coma. Strangely I thought ‘phew she’s safe now’. I just need her to wake tomorrow and start the recovery as we had done so many times before. But she didn’t wake, she wouldn’t come off the breathing machines. They did brain scans to see if any lack of oxygen had given her brain damage.
Isla spent two weeks on life support but when she came around she wasn’t the little girl I knew. She behaved like she was blind and had all over body spasms, they did another brain scan. This time the brain had time to react, they told us she had brain damage. Not from the lack of oxygen but from trauma caused by the intense seizures. They had no other answers for us. They didn’t know what her quality of life would be or if she would survive.
Six weeks in total we spent in The Evelina Hospital.
When we got home our lives had changed. Isla couldn’t see, talk, eat or support herself in any way. She would have muscle spasms that would send her screaming in pain. I was heartbroken and lost. I didn’t know what to do to make things better for her and she couldn’t tell me. We cried for days, weeks and to be honest I still do now, at random times and never in front of Isla. Another mummy said to me ‘you must grieve for the child you lost, and then celebrate the child you have now’.
I must say that took me a very long time. I missed Isla terribly I couldn’t look at photos before the brain damage and if I did I would say ‘mummy misses you so much’.
My heart was full of guilt, guilt for missing my funny energetic Isla. Many children don’t survive what Isla went through and the doctors were surprised she did. I should be grateful for that. But I just couldn’t help it. I missed her terribly. The first year of brain damage has been hard. I’ve fallen in love with my new Isla but still miss the old Isla very much.
Me and my partner struggled at different times. We held each other up when one would break and I’m thankful we are making it through this together.
We have had another little girl who is now 3 months old, Betsy. We were tested before having her and given the all clear. Isla’s Dravet was a spelling mistake in her genes. Such a tiny thing caused devastation in our world. I wish Isla had a better life and didn’t have to fight to survive. However this is what is happening so we have promised to make the most of every day. We lose children with Dravet nearly every month.
Another Dravet mum who recently lost her little boy to SUDEP (sudden unexpected death in epilepsy) said to me ‘it’s the quality of life that’s important not the quantity’.
That’s what we go forward with. Each day is precious with our Isla and I plan to waste no days.
Thanks to The Sandcastle Trust Isla met Santa this year surrounded by all her family.”





