
Our Story
In 2009, at 28 years old Dave, my rugby playing, police officer husband started walking like he was drunk. On a holiday to Spain with friends, he suddenly lost the ability to wear flip flops, they would fly off his feet – it was the running joke of the holiday. Fast forward a few months and he started to have falls, walk with a limp and experience painful spasms in his legs. This was less funny – although we were still not overly concerned, he was young and apparently healthy, we thought perhaps a trapped nerve or a slipped disk was to blame.
What followed was the worst three years of our lives.
A report published by Rare Disease UK found that the average rare disease patient consults with five doctors, receives three misdiagnoses and waits four years for their final diagnosis. This was certainly our experience. Dave was subjected to appointment after appointment where various consultants would shrug their shoulders and an endless round of blood tests, MRIs, lumber punctures and nerve conduction studies. It was made clear to us that it was serious, neurological and degenerative – he wasn’t going to get better, in fact he was going to get a lot worse. Then came a series of misdiagnoses: First MS, then Primary Progressive MS, then Spastic Paraplegia and then PLS (a rare form of Motor Neuron Disease).
It was an awful, lonely time. We felt isolated and lived a day to day life of fear and uncertainty. We were frustrated and terrified.
Finally, in 2012 he received the formal diagnosis of Adrenomyleoneuropathy – a rare genetic neurological disorder. Even the consultant wasn’t expecting to give us a diagnosis that day. We had met him for another routine appointment and a print out of Dave’s latest blood tests which were handed to the consultant by a nurse during the appointment had thrown up the result. He stumbled over his words as he gave us the news and Googled the condition in front of us.
The initial feelings were those of relief. Finally, we knew what was going on, the exhausting battle to kick down every door and shout loud enough to get a definitive diagnosis was over and we could begin to access the help and support Dave desperately needed but was so difficult to obtain without a medical label. But the diagnosis also bought with it the reality of a potentially frightening prognosis and snatched our future away. This wasn’t meant to be part of the life we had planned together – we were newlyweds, had just purchased our first home, both enjoyed our jobs and were planning on travelling, having fun and then perhaps starting a family.
Whilst he was undiagnosed Dave and I would have long conversations where we fantasised about being told it was something temporary, a phase, treatable and would get better or change. That’s hard to do when you have a diagnosis and we went through a long period of grief, mourning the life we thought we were going to have. However, when the shock began to wear off, we entered a period of adjustment and started to look at things more practically. My own ignorance and prejudices surrounding disability initially prevented me from envisioning us ever having a happy family life but you learn to adapt, to assimilate and a future started to emerge albeit most definitely not the one we had planned or hoped for.
Four years on and Dave is spending increasing amounts of time in a wheelchair and lives with the uncertainty that for some people with his condition the prognosis is terminal. Despite the constant daily challenges he faces because of his condition, he still works full time, has become a father to twin boys and enjoys family life.
As time went on we began to learn more about Dave’s condition. He had a less aggressive adult onset form of a gene mutation that usually kills boys before the age of ten. The knowledge that he may not have had a childhood and the opportunity to have a family, made Dave determined not to be overcome with fear about what might happen in the future, to make the best of each day – focus on the happy moments and not let disability be a barrier to experiencing everything that life has to offer and that included enjoying time together as a family.
This however, proved easier said than done. We quickly realised that Dave’s condition would mean we would have to navigate all manner of physical challenges when planning days out or holidays for our family. We began to think about how much harder it must be for other families in the rare genetic condition community to access much needed fun and respite – those that were unable to continue working following theirs of their child’s diagnosis, or those that have several children with the same diagnosis, for example.
That is where the idea of the Sandcastle Trust comes from. The physical emotional and financial strain of having a loved one with a rare genetic condition can often tear families apart. We hope to help other families like ours access holistic respite improving health, wellbeing, happiness and quality of life.

Louie’s Story
It was at five-months-old that Mum Natalie McDougall started to worry about her first-born son Louie.
“He was born very small, just 4lb 11oz, but at five-months-old his foot had dropped into a pointed position,” Natalie, recalls. “He wasn’t meeting milestones, wasn’t sitting up or bearing any weight on his legs.”
Louie was referred to a paediatrician and diagnosed with hypotonia, which is low muscle tone. He had a cast fitted to correct the foot drop and started further tests to determine what could be causing it.
Doctors initially suspected spinal muscular atrophy, but the test came back negative when Louie was seven-months-old. Then in December 2012, Louie, by now aged eight-months-old, became very unwell.
Natalie, from Mitcham, Surrey, says: “We were out shopping when Louie collapsed and stopped breathing. He was rushed to hospital where doctors told us he had respiratory bronchiolitis and his diaphragm had become paralysed. It was the last time he was able to breathe for himself and he was ventilated in hospital for the next seven months.”
Louie was transferred to the Evelina London Children’s Hospital and tested for the rare genetic disorder SMARD (spinal muscular atrophy with respiratory distress). The test came back positive in January 2013. SMARD is an inherited neuromuscular degenerative disease that causes infants to lose voluntary muscular functions, such as breathing and swallowing.
One in 50,000 people carry mutations in the IGHMBP2 gene that is linked to SMARD. Natalie and Louie’s father, Reece were both found to be carriers of the faulty gene.
In SMARD, the link between the brain, the spinal cord and muscles is impaired. Muscles can no longer be stimulated, which causes them to waste and it affects every single muscle in the body. SMARD is a life-limiting condition and there is no treatment available.
Natalie explains: “We were devastated. The doctors told us we needed to let Louie go naturally and there was no hope. I didn’t know 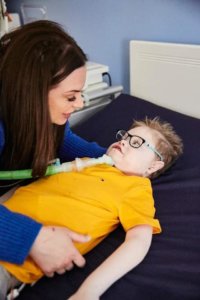 anything about genes or SMARD, but I researched it and pushed for a tracheostomy to be fitted which would enable my baby to come home. In March 2013 Louie had the tracheostomy fitted and was home four months later with a ventilator, which is the size of a lunchbox.”
anything about genes or SMARD, but I researched it and pushed for a tracheostomy to be fitted which would enable my baby to come home. In March 2013 Louie had the tracheostomy fitted and was home four months later with a ventilator, which is the size of a lunchbox.”
At first Louie could still move his limbs, but the condition progressed quickly and he became fully paralysed from the neck down, unable to walk, sit up, lift his head and or speak.
With such complex needs, Louie needed round the clock medical care – which Natalie and Reece were trained to do with some help from night-time nurses.
“We ran our home like an Intensive Care Unit. It was hard work, particularly since we had other children,” Natalie explains. “We both had to give up our jobs to care for Louie. We didn’t want to leave Louie in a hospice.”
Natalie adds: “Despite being under the care of the palliative care doctors Louie was a happy and smiley boy. He loved to be cuddled by his brothers and watch Disney films. He was home schooled by a tutor for one hour a day and used an eye-gaze for simple words.”
 Natalie says: “I was really worried about having other children. We saw a geneticist and whilst the love you have is the same for each child, we decided we wouldn’t continue with a pregnancy if we had another baby with SMARD. I had a CVS diagnostic test with all three pregnancies.
Natalie says: “I was really worried about having other children. We saw a geneticist and whilst the love you have is the same for each child, we decided we wouldn’t continue with a pregnancy if we had another baby with SMARD. I had a CVS diagnostic test with all three pregnancies.
“We attended The Sandcastle Trust Christmas party in 2018,” Natalie explains. “Unfortunately, Louie wasn’t well enough to go, but his younger brothers had a lovely time and it was really helpful to meet other families with rare genetic disorders, share experiences and have support from the charity.”
Then, tragically, on 10 May 2019, aged 7, Louie passed away leaving his family completely devastated.
“Although Louie had a life limiting condition you can never really be prepared to lose a child.” Natalie explains: “When Louie was with us, life was a lot more restricted and our lives revolved around him. Losing him was completely life changing and our whole family found it hard to adjust to our ‘new’ life.”
Louie’s three younger brothers struggled to understand his loss, particularly Charlie. Natalie explains: “Charlie finds Louie dying hard to talk about and will often burst into tears or just say he doesn’t want to talk about it.”
The family was supported by The Sandcastle Trust after Louie’s death. Natalie says: “The Sandcastle Trust arranged for us to have a short break at Butlins. It allowed us a chance to remember Louie and relax whilst creating some fun, happy family memories for Charlie, George and Arthur. Charlie said it was “the best holiday ever!”
Natalie adds: “Louie taught me so much during his life and how to truly love and care for someone. I will miss him every day for the rest of my life.”




Ruairi’s story
Like most 28-year-olds Ruairi Corr wants to live an active life, travel independently and enjoy new experiences. Ruairi loves swimming, hiking, mountain climbing and has even been sky diving – all incredible as Ruairi has the rare genetic condition adrenoleukodystrophy (ALD) and is registered blind, hearing impaired and has epilepsy.
Despite his complex needs, Ruairi, who lives with parents Deirdre, 62, and Aaron, 61, in Exmouth, Devon, was eager to go away on a holiday without his parents. The family applied for the Sandcastle Trust’s programme Sandcastle Memories, which offers respite for families living with a rare genetic condition, in the form of UK breaks and days out.
The Sandcastle Trust was delighted to be able to grant Ruairi a trip to Center Parcs Longleat Forest, where – always accompanied by two carers – he had a fantastic time in the swimming pool, the spa and walking in the forests.
 “Ruairi absolutely loved Center Parcs,” says his mum Deirdre. “He enjoyed the swimming so much and went up and down the rapids repeatedly. He had massages, which relax him and loved the spa and jacuzzi hot tub. He did so much walking his carers kept getting lost, which he thought was very funny. His favourite place to eat was the Pancake House, where he had fabulous pancakes filled with cream and chocolate. He also did an ‘escape room’ activity. It is all brilliant for his fitness and he thrives on being active and busy. We can’t thank the Sandcastle Trust enough.”
“Ruairi absolutely loved Center Parcs,” says his mum Deirdre. “He enjoyed the swimming so much and went up and down the rapids repeatedly. He had massages, which relax him and loved the spa and jacuzzi hot tub. He did so much walking his carers kept getting lost, which he thought was very funny. His favourite place to eat was the Pancake House, where he had fabulous pancakes filled with cream and chocolate. He also did an ‘escape room’ activity. It is all brilliant for his fitness and he thrives on being active and busy. We can’t thank the Sandcastle Trust enough.”
Ruairi’s diagnosis came at the age of five when the family received the devastating news that not only Ruairi, but also his younger brother Tom, who was four, had ALD. ALD is a very rare, inherited genetic condition that causes the build-up of very long chain fatty acids in the brain. This build-up destroys myelin, the protective sheath that surrounds the brain’s neurons, which are the nerve cells that allow us to think and to control our muscles.
Only boys are affected, with mothers often carriers, as was the case for Deirdre, who has mobility difficulties herself. The first sign is that healthy boys start to regress, followed by deafness, blindness, seizures and loss of muscle control. For Ruairi the first signs came around the age of five when he developed processing issues, was diagnosed as being on the autistic spectrum, and was then hospitalised with adrenal problems.
“When the boys were diagnosed we were living in Ireland and we were told it was a terminal illness,” explains Deirdre. “I started researching ALD and found a specialist doctor in Bristol and found out a bone marrow transplant could halt the symptoms. We were initially refused the transplant for Ruairi, when he was six, whereas Tom, then aged five, was at the right stage. We persuaded the doctor to repeat the tests for Ruairi and the consultant agreed to the transplant, whilst warning us the future could be bleak and we had three other young children to look after.”
The bone marrow was donated for transplant by big sister Aisling, who was aged seven. It saved the boys’ lives, but it took 12-18 months to take effect and, in that time, Ruairi lost his sight and his hearing. The treatment was successful for Tom and he is healthy.
When Ruairi was seven the family decided on a major life change, moving to Exmouth so Ruairi could attend a school for the blind in Exeter. Over the years his brother and two sisters went on to live independent lives, something Ruairi found difficult to cope with. With lots of energy and physically fit Ruairi was eager to enjoy new experiences, including going on holiday.
Deirdre says: “Ruairi has carers 24/7 and can’t do anything alone. He says he’d love to be able to walk out the front door on his own and say, ‘see you later’. He is so aware of what is going on and it makes it difficult for him. He thrives on being busy. Ruairi volunteers once a week at a horticultural centre and he has turned our back lawn into a mint garden, which he loves as he has a heightened sense of smell. His carers have become friends of the family; he bakes with one, one carer does woodwork and another is a yoga teacher. He uses an adapted bike to take part in virtual cycles; he has climbed the Three Peaks and Carrauntoohil, the highest mountain in Ireland, a real challenge as streams and rocks are hard when you can’t see. For his 21st birthday he did a sky dive, accompanied by his sister who is a nurse!
him. He thrives on being busy. Ruairi volunteers once a week at a horticultural centre and he has turned our back lawn into a mint garden, which he loves as he has a heightened sense of smell. His carers have become friends of the family; he bakes with one, one carer does woodwork and another is a yoga teacher. He uses an adapted bike to take part in virtual cycles; he has climbed the Three Peaks and Carrauntoohil, the highest mountain in Ireland, a real challenge as streams and rocks are hard when you can’t see. For his 21st birthday he did a sky dive, accompanied by his sister who is a nurse!
“Going to Centre Parcs has had a huge impact on Ruairi’s wellbeing. He came back so happy and wanting to tell us all about it. He gets on so well with his carers – they can have a laugh. The break really lifted his mood and had a positive effect on his mental health. He looks a lot better for the break; brighter, happier and a better colour. It makes him feel like he has an independent life like his siblings.”




Ivan’s story
Three-year-old Ivan Novak was born with the very rare genetic condition Phelan McDermid syndrome (PMS) which affects development, mobility, speech, digestion and shares similarities with autism.
Ivan’s parents Elissa, 32 and Joe, 33, first became concerned when Ivan – their only child – was seven-months-old and was delayed reaching milestones.
 “It was during the first lockdown and no one would see him,” Elissa, from Nuneaton, Warwickshire, recalls. “He then had a big regression around the age of one, following a hand, foot and mouth infection. He lost the words he had, like Momma and Dadda, and his mobility. He regained some skills, although still non-verbal, before regressing again aged two, which is when we got the diagnosis.”
“It was during the first lockdown and no one would see him,” Elissa, from Nuneaton, Warwickshire, recalls. “He then had a big regression around the age of one, following a hand, foot and mouth infection. He lost the words he had, like Momma and Dadda, and his mobility. He regained some skills, although still non-verbal, before regressing again aged two, which is when we got the diagnosis.”
Ivan’s genetic condition affects every area of his life, and he has around ten doctors and practitioners involved in his regular care. He has low muscle tone which affects his fine and gross motor skills, and he is unable to gesture or point. This makes communication difficult for him which is understandably frustrating. He struggles with balance and spatial awareness, which along with low muscle tone causes him to fall and tire quickly. He can walk with assistance.
The biggest problem Ivan faces is with his digestive system. He needs weekly enemas to pass a bowel movement, but these have to be administered in A&E as the funding for community care has been cut. Ivan vomits frequently, which can pose a choking risk and has resulted in weight loss. He now weighs just 11kg. Ivan has needed long stays in hospital and has shown signs of trauma.
Elissa explains: “This last year has been difficult. In Autumn 2022 Ivan spent seven weeks in hospital for pain related to fluid on the brain and sudden seizures.  Then a week after he came home, sadly my Mum passed away. All of a sudden, his grandma was gone from his life and he became very withdrawn. He was having a trauma reaction. He didn’t want to play or do anything at all. All he would do was listen to Waiting On A Miracle, the sad song from the film Encanto.”
Then a week after he came home, sadly my Mum passed away. All of a sudden, his grandma was gone from his life and he became very withdrawn. He was having a trauma reaction. He didn’t want to play or do anything at all. All he would do was listen to Waiting On A Miracle, the sad song from the film Encanto.”
The Sandcastle Trust wanted to help Elissa and Joe create special memories with Ivan through their programme Sandcastle Memories, which offers respite for families living with a rare genetic condition, in the form of UK breaks and days out.
The charity arranged for a week’s stay in March 2023 at the Cowries St Merryn holiday home in Padstow, Cornwall. The break away proved to be a real tonic for Ivan.
After the long drive he went straight upstairs and curled up on a bed with a teddy and fell asleep,” Elissa says. “He was so relaxed in the holiday home. Having not been mobile Ivan started walking on the beaches and he has stayed walking since. One third of Ivan’s life has been spent in pain and distress, with many hospital stays. It is horrible and it is hard to go through, but to see Ivan feeling better and having fun was just brilliant.”
 Whilst in Cornwall the family went on a visit to a farm, Ivan took part in jam tasting and discovered he loved fudge. They particularly enjoyed the beaches, Padstow and relaxing in the house.
Whilst in Cornwall the family went on a visit to a farm, Ivan took part in jam tasting and discovered he loved fudge. They particularly enjoyed the beaches, Padstow and relaxing in the house.
“We all felt a lot better after the break. We spent family time together and it was relaxing. That time together makes a huge difference to our ability to cope with the condition.”







Elijah’s story
Two-year-old Elijah Middleton was born with a rare genetic condition called PTEN hamartoma tumour syndrome (PTEN / PHTS for short). There is no cure or treatment available for this condition, which has wide ranging symptoms and carries an increased risk of certain cancers.
Parents Becky, 30 and Tim, 31, from Carmarthenshire, first noticed Elijah wasn’t meeting his developmental milestones during his eight week check-up. Elijah was born weighing 10lb 1oz and with a very big head and at eight weeks old his muscle tone was too low to support his head. He also had delayed motor development. He was referred to a paediatric consultant where he had a number of tests and assessments and then onto a geneticist who straight away suspected PTEN syndrome. PTEN syndrome is caused by changes in the PTEN gene, which is a tumour suppressor. An alteration in this gene can lead to non-cancerous tumours and increase the lifetime risk of some types of cancerous tumours too. Elijah’s genetic testing came back positive, and the family got a diagnosis in October 2022, when he was 15 months. By then his gross motor skills and speech and language were very delayed.
Becky explains: “After the geneticist explained what PTEN was and the symptoms associated with it, it sounded so much like Elijah. We were almost expecting to hear that the test was positive for PTEN, but it was still a massive shock. We all had blood tests, including our eldest son Asher, who is four, but none of us have the condition. We really don’t know what the future holds as it can cause fatty lumps which can be cancerous. So far Elijah has just had a scan on his abdomen for lumps. He has vomiting episodes which means he loses weight. He always seems to be poorly as his immune system is not great.
“There is a lot of guesswork in looking after Elijah as he doesn’t communicate his needs. He has very few words so can’t tell us if he is hungry or thirsty. He struggles to understand that a ball means we play, or a bath is bath time. He doesn’t always respond to speech, but it is too early for an autism diagnosis.”
Speech and Language have been supporting him and providing strategies to better support Elijah and his family. The family are waiting for a referral to a dermatologist to check Elijah’s skin for lumps that may become cancerous. When he is around seven, Elijah will be monitored for any lumps on the thyroid gland. He has physio for his low muscle tone, balance, co-ordination and hypomobility. During the summer he will have six weeks of hydrotherapy. He is also having hearing tests and waiting to have an overnight pulse oximetry sleep study.
Becky says: “The diagnosis period has been very stressful on family life. Tim now works at home, he’s a Learning and Development Advisor, so he can help when he can, but it’s not easy. I’m with Elijah 24/7. I have to get in the bath with him as his muscle tone is too low to sit safely. He slumps when he is eating and bathing. We are waiting for a specialist highchair and bath chair which will make a big difference. My family are an hour away and Tim’s family are in Shropshire, so we have no family close by. Going out and doing normal family activities are so difficult for us. We hadn’t been on a holiday together with the children due to Covid and then it just seemed too scary and daunting.”
The couple applied for the Sandcastle Trust’s programme Sandcastle Memories, which offers respite for families living with a rare genetic condition, in the form of UK breaks and days out.
The Sandcastle Trust wanted to help Becky and Tim create special memories with Elijah and Asher and were able to arrange a week’s stay in May at Calcite Cabin, Trevassack Lake in Cornwall.
“I broke down in tears when I heard we had been granted the holiday,” Becky says. “After all the assessment and diagnosis, we had gone through a rollercoaster of emotions, for the Sandcastle Trust to say you can get away, have a break from the hospital appointments and take a step back and just be a family, was such a blessing for us all.”
“The house was perfect. It was gated off and Elijah could crawl around safely. We went to soft play, an aquarium and visited a farm. The beach was so close we were back and forth to it every day. It was nice to see Elijah’s face light up. We also walked around the Lizard and took a trip to Newquay. The area is beautiful and so peaceful. Then once bedtime hit Tim and I chilled out in the hot tub.”
Becky adds: “The boys loved the holiday and for us it helped us to relax and was great to give our minds some respite. We had been feeling a lot of anxiety about what the future would be like but the week in Cornwall gave us a breather from our everyday life, it changes your outlook and your ability to cope with the condition. It gave us confidence, look what we could do in a week! We can’t thank the Sandcastle Trust enough.
“Amazingly Elijah tried to stand up in the sand which was really exciting for us as he wasn’t standing independently before. He seemed so much more engaged and enjoyed spending time as a whole family. It was such a great week!”

Tabby’s Story
Meet 8 year old Tabby.
 Tabby’s family have been on a very long journey to obtain the correct diagnosis for her and she has recently been diagnosed via the 100,000 Genomes Project with TANGO2, a very rare genetic metabolic condition that causes episodes of metabolic crisis and carries the risk of cardiac failure. It is thought that there are less than 30 affected individuals worldwide. Tabby also has a chromosome disorder – 22q11 deletion. This affects her in various ways; she has learning difficulties, speech difficulties and a cleft palate.
Tabby’s family have been on a very long journey to obtain the correct diagnosis for her and she has recently been diagnosed via the 100,000 Genomes Project with TANGO2, a very rare genetic metabolic condition that causes episodes of metabolic crisis and carries the risk of cardiac failure. It is thought that there are less than 30 affected individuals worldwide. Tabby also has a chromosome disorder – 22q11 deletion. This affects her in various ways; she has learning difficulties, speech difficulties and a cleft palate.
Hannah, Tabby’s mum explains “It has been a scary time for us as a family getting our head around the TANGO2 diagnosis and it has been made so much harder by the coronavirus lockdowns and the isolation this brings.”
Hannah got in touch with The Sandcastle Trust and told us how Tabby and consequently the whole family have really struggled with the Covid-19 restrictions, “Tabby does not really play with toys, she needs 1:1 supervision and does not play independently. She hates being cooped up indoors and loves to be outside whatever the weather. She has found this winter lockdown particularly hard.”
Tabby’s family were one of the first families to benefit from our new category of support – ‘Sandcastle Memories At Home’ and we were pleased to provide Tabby with an Artmark House. A fun alternative to easels, the Artmark House will provide Tabby with her own sheltered space with white board, black board and perspex surfaces to draw and paint on.

The Artmark House is in the garden which is where Tabby is at her happiest. It gives her a safe place where she can be creative and try to develop more independent play. When she first saw the house she was so excited. I have not heard her laugh this much in ages. She loves painting and drawing in it. Thank you so much to The Sandcastle Trust.




Sam’s Story
Three-year-old Sam Murray has just been on his first holiday to Cornwall. He watched in awe as waves crashed against the rocks, loved building sandcastles with his dad and saw lifeboats setting off on a rescue. It sounds like just a normal family holiday, but for Sam and his parents Harriette and Graham, it was an extraordinary holiday and one they didn’t think they would be able to go on.
For Sam has a rare and life-limiting genetic condition, which causes damage to the muscles throughout the body. And this holiday was a respite break arranged by The Sandcastle Trust for families living with a rare genetic condition.
Harriette and Graham drove the six hours from their home in Kidderminster, Worcestershire, with the car full of all the equipment needed to look after Sam, including his wheelchair, night-time ventilator, peg feeding equipment, medication, and rescue oxygen. It was the first time the couple felt confident enough taking Sam away for a week, due to his complex medical needs.
feeding equipment, medication, and rescue oxygen. It was the first time the couple felt confident enough taking Sam away for a week, due to his complex medical needs.
Harriette says: “Sam loved it. He has never really seen the sea before, and he had never been to a beach. The weather was rough, and he loved watching the waves crashing against the rocks. He just wanted to stare at the sea for an hour. He thought it was brilliant. For us the trip allowed us to have that break from reality and spend quality time together.”
In February 2021, aged 13 months, Sam was diagnosed with infantile-onset Pompe Disease, and was found to be in cardiac and respiratory failure. Doctors told parents Harriette Cooper, 30, and Graham Murray, 40, they didn’t think Sam would survive.
“It was heart-breaking, and our lives started unravelling before us,” Harriette recalls. “You never think this will happen to you. We first noticed at around seven months that Sam’s arms were floppy. But because of the pandemic there were no baby weigh-ins or health visitor appointments. We asked the health visitor to see him when he was ten months old and then he was referred to hospital.
“The silver lining was that the pandemic lockdown had kept him safe and away from germs, but when we got the diagnosis we couldn’t even hug our families. It made it tremendously difficult.”
Pompe disease is a metabolic genetic condition in which complex sugar called glycogen builds up in the body’s cells, as a result of a deficiency in an enzyme that breaks down the sugar. This build-up of glycogen affects all muscles of the body, including the heart and lungs. Without treatment the prognosis is bleak.
Luckily Sam was approved for a new experimental treatment of enzyme therapy which started as a trial before being licensed in August 2022. After four months in hospital and two weeks in a hospice to support the couple in caring for Sam at home, he was able to go home. His parents were trained to give the enzyme replacement therapy infusion, every fortnight, at home.
Since then Sam has defied the doctors’ expectations. Sam has regained some movement in his arms and upper body and improved his core strength, so he can sit with support and play with toys, greatly improving his quality of life. He has many complex medical needs; he is fed through a gastrostomy tube and he needs to use a wheelchair. He sleeps with a ventilator as he can stop breathing during the night and has an overnight carer five nights a week. However, he no longer needs 24-hour oxygen, which is a real breakthrough.
Harriette says: “Sam’s condition will always be life-limiting, but doctors are thrilled with how he is doing. Sam attends an early year’s nursery and a special needs school for one day a week and is able to play with toys – he especially loves trains and Toy Story. He is non-verbal but he babbles and uses Makaton. You can definitely tell what he wants!”
With Sam no longer needing full-time oxygen, the couple started to think about the possibility of a holiday together and applied for the Sandcastle Trust’s programme Sandcastle Memories, which offers respite for families living with a rare genetic condition, in the form of UK breaks and days out.
 The Sandcastle Trust wanted to help Harriette and Graham to create special memories with Sam and were able to arrange a week’s stay at Waves End cottage in Polzeath, Cornwall.
The Sandcastle Trust wanted to help Harriette and Graham to create special memories with Sam and were able to arrange a week’s stay at Waves End cottage in Polzeath, Cornwall.
Harriette used to go to Cornwall every year on family holidays and had many happy memories of her trips as well as one very difficult memory.
“The last time we went to Cornwall was six years ago,” she explains. “We suffered a miscarriage there and it was a bit of a traumatic experience. We kept a bit of Cornwall in our hearts, so it was emotional. I was nervous to take Sam down there, with his medical needs, but it was nice we could share Cornwall with him and heal.”
Sam had a fantastic time on his first proper holiday and the whole family had a chance to relax and recharge.
Harriette says: “He was able to build sandcastles with his dad and we carried him into the sea to get his feet wet. When we got home, we filled a sandpit as he wanted to play with the sand. We walked coastal paths, visited Polzeath, we went to the Eden project which was amazing and so sensory and accessible for Sam. We all thought it was fantastic and Sam loved the waterfall. We went to an accessible, sensory orchard and playground at Pentire Point National Trust and on Mother’s Day we went for a cream tea. Then in Padstow Sam loved seeing the boats in the harbour and saw lifeboats launching. It was Graham’s 40th birthday just before we went so it was great to mark that too.”
“It was lovely to get away and do something just for us,” Harriette adds. “It is a full-time job and exhausting looking after a child with medically complex needs. In Cornwall we could switch off from the day to day, press pause and allow us to be a normal family. We are so grateful to the Sandcastle Trust.”










Eddie’s Story
Eddie Braun is a loud, sometimes cheeky four-year-old who is determined to let his mum know exactly what he wants.
His default setting is happiness, despite the numerous challenges he faces. He absolutely loves cycling and swimming.
Eddie has an ultra-rare genetic condition caused by the genetic mutation COL4A2 which is linked to small blood vessel disease. For Eddie this means he had at least one bleed on the brain before he was even born, resulting in brain damage. This in turn has meant he is registered as severely sight impaired, he has cerebral palsy, global developmental delay and complex epilepsy.
The Sandcastle Trust awarded the Braun family a holiday at Bluestone, a holiday park, in Pembrokeshire, Wales, for a week.
Eddie’s mum, Ilmarie, says: “It was one of the first times we have been away just the four of us – just us as a family. I’m sure without The Sandcastle Trust we would never have done it.
“We wouldn’t have been able to budget for it. Financially it is difficult because I’m not working. And also because increasingly there aren’t many things we can do easily as a family.
“I have to be available to take care of Eddie and to get him to all of his appointments. In one week alone he had nine appointments with the NHS.
“There have been periods where Eddie has had 10 clusters of spasms (seizures) a day. At the moment I can’t work because if Eddie has a bad run of seizures, he needs to go to hospital. It took us a long time but thankfully we have got better seizure control now, although he still has seizures every day.”
Ilmarie, 42, explains: “It was really easy to apply to the Sandcastle Trust and they were so lovely to deal with. The whole process removed any stress – which is brilliant when you are looking after a profoundly disabled child.”
Ilmarie, who is married to Alex Braun, also 42, a builder, lives in Chester with Eddie and his older brother, Thomas, eight years old.
She continues: “What was so special about the week The Sandcastle Trust gave us was our two sons got to share a bedroom and enjoy activities together.
“Thomas absolutely adores Eddie, but most of the time in order for him to enjoy anything resembling what other eight-year-olds do, one of us has to take him away and the other parent stays with Eddie. Our little family is split in two most of the time as a result.
“Thomas’s whole life has been impacted since Eddie was born. It is very hard for the siblings of disabled children.
 “We never thought we’d be able to have the boys sharing a room and just to be able to go out on the bikes together in a safe environment and head to the swimming pool – sometimes more than once a day was incredible.”
“We never thought we’d be able to have the boys sharing a room and just to be able to go out on the bikes together in a safe environment and head to the swimming pool – sometimes more than once a day was incredible.”
Eddie is profoundly disabled and while he can feed himself, his food needs to be cut up for him.
Ilmarie continues: “Bluestone is so well organized we even managed to eat out twice. It was such a treat not to have to think about the shopping, cooking and clearing up.
“The trip was perfect. It allowed us to reconnect as a family and have a little bit of a breather too.
“It has given Alex and I the confidence to feel we could do the same kind of trip again.
“For any other families out there who are in desperate need of a break. Have a look at The Sandcastle Trust. They helped us do the normal things that other families do and gave us a chance to spend quality time together again.”

Kirsten’s story
It’s not just families with young children to benefit from The Sandcastle Trust’s accessible holiday breaks. Families with adult children with genetic conditions can also apply to our Sandcastle Memories programme, and benefit from a day out with their family, or a fully accessible UK holiday.
These respite breaks are invaluable for families such as the Hardmans – who are caring full time for their daughter Kirsten, 27, who has a genetic condition and lives with complex needs and mobility issues. Kirsten needs round the clock care as she can injure herself, she is a wheelchair user, has developmental delay, scoliosis, autism, is doubly incontinent and is non-verbal.

Kirsten was born in South Africa, and at birth had struggled to feed and ‘failed to thrive’. The family moved to Liverpool when Kirsten was 15 and she settled into a new school. Despite genetic testing in South Africa, the family did not have a diagnosis. Then in 2019, a breakthrough came from the Deciphering Developmental Disorders (DDD) project – a diagnosis of Shaaf-Yang Syndrome (SYS), a rare genetic condition which shares similarities with Prader Willi Syndrome. Children with SYS can have low muscle tone, feeding difficulties, developmental delay and autism. They can also have weight issues and feel hungry all the time.
Jane Hardman, 54, is a full-time carer for Kirsten. Jane’s husband Bruce, 53, is a logistics driver and their eldest daughter Caitlyn, 29, lives nearby.
“It is a rare condition and there are only about 3000 cases of SYS worldwide,” Jane explains. “When Kirsten was born, she had difficulty feeding and she was tube fed for the first six months of her life. She stopped breathing a lot. She couldn’t open her hands; she had club foot and a bad squint in her eye that was operated on. The doctors said she wouldn’t walk and gave her six months to live. She actually did walk, aged three-and-a-half.
“It was costing everything we had to stay in South Africa and pay for medical bills. One doctor said, ‘I’m wasting your money.’ There was nothing they could do and didn’t know what the cause was. We came back to the UK – we had family here – and joined the new DDD study. Kirsten, Bruce and I had swabs taken and they managed to find exactly what it was, which was amazing. It really helps with understanding her needs.”
Kirsten attends a day centre two days a week, but an assistant is only available to attend with Kirsten on one day, so Jane goes with her on the other day. Kirsten uses a wheelchair as she can’t walk for long distances due to low muscle tone and scoliosis. She needs incontinence pads and also has sleep apnoea and can stop breathing at night. She is non-verbal – communication is difficult as she won’t use online communication apps, but she understands everything. She likes Lego, she’s now working on 8+ years, jigsaws and computers, but her favourite thing to do is swim.
Jane says: “I look after Kirsten full-time and we haven’t had a holiday for a long while. During the pandemic, the day centres were closed, so Kirsten was at home full time. It was one of our social workers who asked if I need a break, so I applied to The Sandcastle Trust.
“Kirsten loves the pool, she’ll be in it for four hours and she’s happy as anything. She loves to go anywhere with a pool so we were thrilled to get the Skegness caravan break with a pool on site!”
 The Hardmans stayed at The Sandcastle Trust’s fully adapted static caravan in Skegness this summer (2023) and were joined by Kirsten’s sister Caitlyn to help out.
The Hardmans stayed at The Sandcastle Trust’s fully adapted static caravan in Skegness this summer (2023) and were joined by Kirsten’s sister Caitlyn to help out.
Jane explains: “Kirsten loves caravans, she doesn’t want to go on any other holidays. She loves the penny arcades, the parks and the swimming pool. She slept with me so I can watch her and the bed was so comfortable. We get nervous in the night as not only does she have sleep apnoea, but she has been known to get up to mischief, she flooded my bedroom at home once.
“Every day we went out and fed the ducks and Kirsten would go swimming in the afternoon and then go to the play area or the beach front. One day we went to the Seal sanctuary, which was great fun, she loved getting up close to the seals. They jumped up and she got nice and wet – she loved that!”
For the Hardman family, the Skegness holiday gave them precious family time together with memories to cherish.
Jane says: “It was just great to make memories with Kirsten. It makes your day to see the smile on her face. She was forgotten about in South Africa and now she lives a life and she’s happy. It’s all I wanted for her.”

Maddison’s Story
Feisty, confident Maddison Sherwood dreams of being an actress.
The ten-year-old student goes to drama school in her spare time and loves nothing more than singing and dancing.
 Maddison has the rare genetic disorder Spinal Muscular Atrophy with Respiratory Distress (SMARD) which means she uses a ventilator and tracheotomy to help her breathe and a wheelchair to get around. Her parents have become 24-hour carers.
Maddison has the rare genetic disorder Spinal Muscular Atrophy with Respiratory Distress (SMARD) which means she uses a ventilator and tracheotomy to help her breathe and a wheelchair to get around. Her parents have become 24-hour carers.
The Sandcastle Trust took great pleasure in organising and funding a trip for Maddison’s family to London’s West End to see The Lion King.
Maddison’s mum Lidia, 32, says: “It was the most wonderful couple of days which we will always remember. It was time away when could we feel like a “normal” family – we could forget the medical side of our lives and the constant fighting for Maddison.
“We felt so grateful the children could experience London and have fun.”
Lidia explains: “After I contacted the Sandcastle Trust they organised our accessible hotel next to the theatre, tickets for the show and made sure both were accessible for Maddison’s wheelchair. They paid for an overnight carer so Maddison’s dad and I could sleep and even ensured we could park our car near the hotel.
“It was just brilliant. Maddison absolutely loved the show.”
Maddison is Lidia and her husband Jamie’s fourth child. Their other children are Lacey, 17, Harley, 14, and Jayden, 11.
Lidia says: “She wasn’t planned but she was very much wanted. It wasn’t until she was six months old that we realised something was wrong. She just caught a cold but was having real difficulty breathing. We took her to Queen’s Hospital in Nottingham and didn’t come out for 11 months.
“She went into respiratory distress and her heart rate was over 200 bpm. That was February 2009, our lives were never going to be the same again.”
Doctors tested Maddison for different disorders and were about to leave her undiagnosed when someone suggested testing for SMARD. Lidia says: “They told us what it was and said she wouldn’t live past two, until then she would just be a pair of eyes. At 2am they suggested we turn off the ventilator that was keeping Maddison alive.
“But we felt that where there was life there was hope. We kept fighting for her.”
Now when those doctors see Maddison they tell Lidia how glad they are the couple made that decision. Maddison goes to mainstream school and is about to take her SATs, she even presents her own YouTube videos.
Life has not been easy.
 Lidia recalls: “We spent so much time in hospital I missed lots of Jayden’s milestones, like him taking his first steps. He still suffers from separation anxiety. People don’t think of the impact on siblings.
Lidia recalls: “We spent so much time in hospital I missed lots of Jayden’s milestones, like him taking his first steps. He still suffers from separation anxiety. People don’t think of the impact on siblings.
“The hospital would not let Maddison home until we were trained in her care. Jamie had to give up his job as a builder to look after her with me.”
Lidia adds: “Money is always tight and the other three children have had to become young carers. They are fantastic around Maddison but they often have to come second or miss out on things like football training. We can never go to parents evening together let alone go for a night out. These are such simple things other people take for granted.
“We get some help but it is not very consistent, paid carers often let us down.
“To be able to travel to London and forget all of that is something we will remember forever.”






